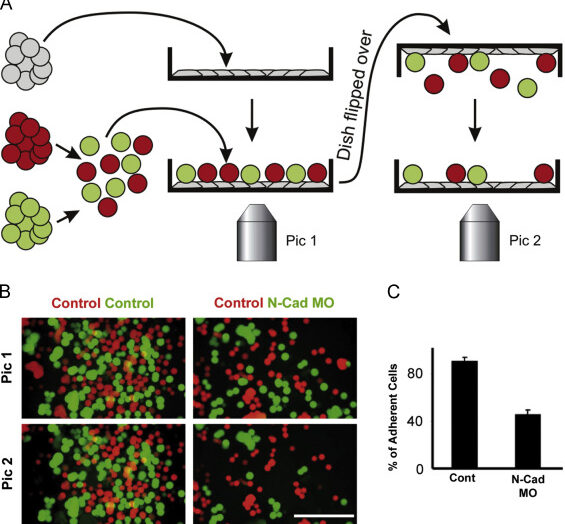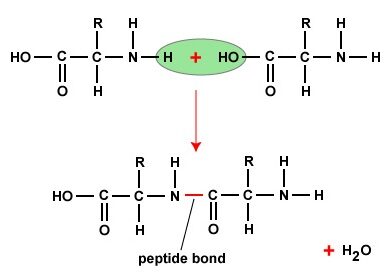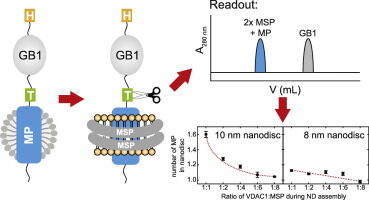
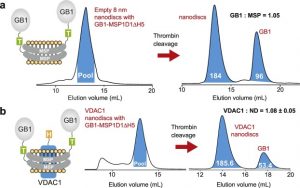
Description
Recent studies on G protein-coupled receptors revealed that they can dimerize. However, the role of each subunit in the activation process remains unclear. The receptor for γ-amino-n-butyric acid type B (GABAB) is composed of two subunits: GB1 and GB2. Both consist of an extracellular domain (ECD) and a heptahelical domain composed of seven transmembrane α-helices, loops, and the C-terminus (HD). While GB1 ECD plays a critical role in ligand binding, GB2 is required not only for targeting the GB1 subunit to the cell surface, but also for receptor activation. Here, by analyzing chimeric GB subunits, we show that only GB2 HD contains the necessary determinants for G-protein signalling.
However, GB1 HD improves docking efficiency. In contrast, although the ECD of GB1 is sufficient to bind GABAB ligands, the ECD of GB2 increases the affinity of the agonist at N-terminal 6xHis-GB1-tagged Recombinant and is required for receptor activation by the agonist. These data indicate that multiple allosteric interactions between the two subunits are required for wild-type functioning of the GABAB receptor and further highlight the importance of the dimerization process in GPCR activation.
Materials and methods
- Construction of chimeric receptors
The N-terminal c-Myc and HA-tagged GB1a and GB2 coding sequences described previously (Pagano et al., 2001) have been subcloned into the expression vector pRK5. To construct the GB1/2 and GB2/1 chimeric receptors, a unique Bsu36I restriction site was introduced into the region encoding the tripeptide Pro434–Pro435–Ala436 and Pro405–Pro406–Lys407 located at the C-terminus of the extracellular sequence. of the c-Myc- or HA-GB1a and GB2 subunits, respectively. The fragment between the EcoRI site preceding the start codon and the introduced Bsu36I site was exchanged between the two plasmids, thus creating the plasmids expressing GB1/2 and GB2/1 chimaeras. Construction of the HA-mGlu5 receptor expression vector has been previously described (Ango et al., 1999).
- Cell culture, immunofluorescence and expression in HEK 293 cells
Human embryonic kidney (HEK) 293 cells were cultured and transfected by electroporation as previously described (Franek et al., 1999). After electroporation, cells were plated on polyornithine-coated plates. Serum, culture media and other solutions used for cell culture were from Life Technologies, SARL (Cergy Pontoise, France). Immunofluorescence on intact cells using the c-Myc or HA antibodies was performed as previously described (Pagano et al., 2001).
- Ligand binding assay
Ligand binding experiments were performed on intact HEK 293 cells seeded in 24-well plates just after electroporation (10 7 cells per plate). The next day, cells were equilibrated at 4 °C, washed three times with ice-cold binding buffer (20 mM Tris-HCl, pH 7.4, 118 mM NaCl, 1.2 mM KH2PO4, 1.2 mM MgSO4). mM, 4.7 mM KCl and 1.8 mM CaCl2) and incubated in the presence of 0.1 nM [125I]CGP64213 with or without unlabeled ligands at the indicated concentration (200 mL final volume, for 3 h at 4°C).
The incubation was terminated by washing three times with ice-cold binding buffer. Cells were then disrupted with 400 ml 0.1 M NaOH and bound radioactivity was counted. The amount of protein in each well was measured by the Bradford method. Nonspecific binding was determined in the presence of 1 mM GABA. The concentration of [125I]CGP64213 used in the displacement experiments (0.1 nM) was approximately 10-fold lower than the affinity of this radioligand on GB1.
Ki values were calculated according to the equation IC50 = Ki (1 + [L]/Kd); Kd was assumed to be equal to Ki in the case of CGP64213. GABA was obtained from Sigma (L’Isle d’Abeau, France). [125I]CGP64213 was synthesized as described elsewhere (Kaufmann et al., 1997a) and labelled with a specific radioactivity of >2000 Ci/mmol (ANAWA AG, Wangen, Switzerland). L-baclofen was synthesized in the research laboratories of Novartis Pharma in Basel (Froestl et al., 1995). Displacement curves were fitted as previously described (Galvez et al., 2000a).
- Western transfer
Forty-eight hours after transfection, cells were washed twice in ice-cold Tris-Krebs buffer containing protease inhibitors (Roche Diagnostics, Meylan, France), discarded, and centrifuged to collect membranes. Samples (10 μg) were loaded onto a Tricine-SDS gel for electrophoresis and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Orsay, France). After incubation in phosphate-buffered saline (PBS)/5% milk, membranes were incubated with anti-HA monoclonal antibody (1/3000; Roche Diagnostics) at room temperature for 2 h. After washing, the membranes were incubated overnight at 4°C with an anti-mouse HRP antibody (Roche Diagnostics). The signal was revealed using an ECL chemiluminescent assay (Amersham Pharmacia Biotech, Orsay, France).
- Photoaffinity labelling and co-immunoprecipitations
Crude membranes of HEK 293 cells expressing the different combinations of receptors were prepared and labelled with the specific photoaffinity ligand GB1 ECD [ 125 I]CGP71872 as previously described (Kaufmann et al., 1997b). Briefly, 24 h after transfection, cells were washed, homogenized in cold Tris-Krebs buffer (20 mM Tris-HCl, pH 7.4, 118 mM NaCl, 5.6 mM glucose, 1.2 mM KH2PO4, MgSO4 1.2 mM, 4.7 mM KCl and 1.8 mM KCl). CaCl2), and centrifuged for 20 min at 40,000 g.
The pellet was resuspended in Tris-Krebs buffer. Membranes (50 µg protein) were incubated in the dark for 1 hour at room temperature with 0.8 nM [125I]CGP71872. Nonspecific binding was determined in the presence of 1 mM CGP54626A. Binding was stopped by two washes with cold Tris-Krebs buffer prior to UV cross-linking. For immunoprecipitation, labelled membranes (200 mg) were resuspended in 100 mL of buffer [50 mM HEPES pH 7.5, 150 mM NaCl, 1% (w/v) deoxycholate, and protease inhibitors] at 4 °C. and then centrifuged at 100,000 g for 1 hour.
After a 5-fold dilution in protein buffer [20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% Triton X-100, protease inhibitors ( Roche Diagnostics, Meylan, France)], supernatants were incubated overnight at 4°C with anti-HA monoclonal antibody (clone 12CA5, 1/250; Roche Diagnostics) and protein A-Sepharose. The beads were washed five times with cold 0.1% Triton X-100 protein buffer. Radioactivity associated with the pellet was counted and labelled immunoprecipitated receptors were detected by SDS-PAGE and autoradiography.
- Determination of accumulation of inositol phosphate (IP)
HEK 293 cells were transfected as described above with the receptor constructs in pRK5 (2 mg), Gqi9 expression vector (2 mg), and carrier DNA (pRK6, 4 mg) (Franek et al., 1999). Determination of IP accumulation was performed 15 h after transfection as described elsewhere (Franek et al., 1999).